| Linia 51: | Linia 51: | ||
Wiele dziedzicznych zespołów nowotworowych (np. stwardnienie guzowate), ma swoją genezę w mutacjach genów dla negatywnych regulatorów ścieżek zależnych od mTOR, jak np.: TSC1 (Tuberous Sclerosis 1), TSC2 (Tuberous Sclerosis 2), LKB1 (liver kinase B1, znany również jako STK11, Peutz-Jeghers syndrome), PTEN (phosphatase and tensin homolog deleted on chromosome ten, znany róenież jako MMAC1, mutated in multiple advanced cancers 1), NF1 (Neurofibromatosis) czy VHL (Von-Hippel Lindau). Onkogenne mutacje genów sieci sygnalnej PI3K-AKT, w sposób bezpośredni powiązanej z kompleksami TROC1 oraz TROC2 mogą również i często powodują rozwój nowotworów. | Wiele dziedzicznych zespołów nowotworowych (np. stwardnienie guzowate), ma swoją genezę w mutacjach genów dla negatywnych regulatorów ścieżek zależnych od mTOR, jak np.: TSC1 (Tuberous Sclerosis 1), TSC2 (Tuberous Sclerosis 2), LKB1 (liver kinase B1, znany również jako STK11, Peutz-Jeghers syndrome), PTEN (phosphatase and tensin homolog deleted on chromosome ten, znany róenież jako MMAC1, mutated in multiple advanced cancers 1), NF1 (Neurofibromatosis) czy VHL (Von-Hippel Lindau). Onkogenne mutacje genów sieci sygnalnej PI3K-AKT, w sposób bezpośredni powiązanej z kompleksami TROC1 oraz TROC2 mogą również i często powodują rozwój nowotworów. | ||
| − | + | ||
{|class="wikitable" style="text-align:center; border="1" | {|class="wikitable" style="text-align:center; border="1" | ||
|+Schematy ścieżek sygnalnych, w które zaangażowana jest kinaza mTOR | |+Schematy ścieżek sygnalnych, w które zaangażowana jest kinaza mTOR | ||
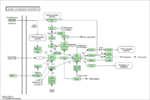
!width=200px|[[File:mTOR-Rysunek3.png|thumb|center|150px|link=http://www.kegg.jp/kegg-bin/highlight_pathway?scale=1.0&map=hsa04150|baza KEGG]] | !width=200px|[[File:mTOR-Rysunek3.png|thumb|center|150px|link=http://www.kegg.jp/kegg-bin/highlight_pathway?scale=1.0&map=hsa04150|baza KEGG]] | ||
!width=200px|[[File:mTOR-Rysunek4.gif|thumb|center|130px|link=http://jcs.biologists.org/content/122/20/3589/F1.poster.jpg|źródło: {{r|Laplante and Sabatini 2009a}}]] | !width=200px|[[File:mTOR-Rysunek4.gif|thumb|center|130px|link=http://jcs.biologists.org/content/122/20/3589/F1.poster.jpg|źródło: {{r|Laplante and Sabatini 2009a}}]] | ||
| − | !width=200px|[[File:mTOR-Rysunek5.gif| | + | !width=200px|[[File:mTOR-Rysunek5.gif|thumbnail|center|link=http://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=3281571_cshperspect-SIG-011593_F2.jpg|źródło: {{r|Laplante and Sabatini 2012}}]] |
|} | |} | ||
| − | + | ||
| + | |||
'''Zobacz też''' | '''Zobacz też''' | ||
Wersja z 11:18, 28 maj 2014
Kinaza mTOR
mTOR (mechanistic target of rapamycin, mammalian target of rapamycin) jest kinazą białkową serynowo-treoninową (EC 2.7.11.1) zdolną do wiązania kompleksu rapamycyny z białkiem FKBP12 (FK506-binding protein 12).[1][2][3] Stąd wywodzi się inna nazwa mTOR - FRAP1 (FKBP12-rapamycin complex associated protein 1). Należy do rodziny kinaz PIKK - kinaz spokrewnionych z kinazą fosfatydyloinozytolu (phosphatidylinositol 3-kinase-related kinase). Jest białkiem wielodomenowym o ciężarze cząsteczkowym 288.9 kDa. Od N-końca wyróżnić można następujące domeny: HEAT (Huntingtin-EF2-A subunit of PP2A-TOR1, aminokwasy 1-1512), FAT (FRAP-ATM-TRAP, 1513-1910), FRB (FKBP12 rapamycin binding, 2015-2114), domena kinazowa (2181-2431), NR (negative regulatory) oraz FATC (FRAP-ATM-TRAP C-terminal, 2517-2549). HEAT jest domeną zbudowaną z powtórzeń α-helis tworzących prawoskrętną superhelisę (jest to tzw. armdillo-like fold). Rozbudowana powierzchnia domeny HEAT umożliwia efektywne oddziaływanie z cząsteczkami makromolekuł białek czy kwasów nukleinowych. Domena FRB tworzy α-helikalną strukturę wiążącą rapamycynę. Domena katalityczna kinazy mTOR jest funkcjonalną białkową kinazą serynowo/treoninową. Jej sekwencja wykazuje wysoki stopień podobieństwa do kinaz fosfatydyloinozytolu.
| TORC1 | TORC2 |
|---|---|
| mTOR | |
| MLST8 | |
| DEPTOR | |
| RAPTOR | RICTOR |
| PRAS40 | PROTOR1 |
| RHEB | mSIN1 |
Kinaza mTOR współtworzy w komórce dwa kompleksy białkowe o odmiennych funkcjach: TORC1 oraz TORC2.[4][2][3] Jedynie pierwszy z nich jest hamowany przez rapamycynę. TORC1 integruje ścieżki sygnałowe kontrolujące wzrost, proliferację czy metabolizm komórki w odpowiedzi na sygnały pochodzące od hormonów, czynników wzrostu, składników odżywczych (aminokwasów), statutu energetycznego komórki, tlenu, a także stresu genotoksycznego. Aktywacja TORC1 polega na fosforylacji czynników wymiany nukleotydów guaninowych: TSC1 (hamartyna) oraz TSC2 (tuberyna), co prowadzi do aktywacji GTPazy RHEB, a w konsekwencji - wyłączenia inhibicji kompleksu TORC1 i jego aktywacji. Fosforylacja kinazy mTOR na Ser1261 stymuluje jej aktywność autofosforylacyjną. Fosforylacja ta jest zależna od ścieżki insulinowej i obecności aminokwasów. Aktywacja TORC1 przez aminokwasy wymaga przemieszczenia kompleksu do lizosomów, zależnego od GTPaz RAG (RRAGA, RRAGB, RRAGC, RRAGD). TORC1 jest hamowany w warunkach niskiego poziomu energetycznego komórki, a także hipoksji poprzez aktywację PRKAA1 i REDD1.
Najważniejsze procesy regulowane przez TORC1 to:
- biosynteza białek - poprzez fosforylowanie takich regulatorów translacji, jak: 4E-BP1 (eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1) oraz S6K1 (S6 kinase 1)[5];
- biosynteza lipidów - poprzez fosforylowanie czynników transkrypcyjnych SREBP1/2 (sterol regulatory element-binding protein 1/2) oraz PPARγ[6];
- produkcja ATP i ekspresja genów enzymów glikolitycznych - poprzez aktywację czynnika HIF1α[7];
- angiogeneza - poprzez aktywację HIF1α i ekspresję VEGF (vascular endothelial growth factor)[8];
- autofagia (regulacja negatywna) - poprzez fosforylowanie i supresję kompleksu ULK1/Atg13/FIP200 (unc-51-like kinase 1/mammalian autophagy-related gene 13/focal adhesion kinase family-interacting protein of 200 kDa)[9];
- tworzenie lizosomów - poprzez czynnik transkrypcyjny TFEB i ekspresję genów białek lizosomalnych[10].
TORC2 reguluje przeżycie komórki, metabolizm, a także organizację cytoszkieletu poprzez fosforylowanie takich kinaz z grupy AGC, jak: AKT, SGK1, PKCα.[2][11]

Znaczenie kliniczne kinazy mTOR
Ścieżki sygnałowe, w które zaangażowana jest kinaza mTOR, kontrolują różnorakie procesy składające się na wzrost czy proliferację komórek. Są to m.in.: angiogeneza, adipogeneza, aktywacja limfocytów T. Aktywacja ścieżek sygnalnych mTOR sprzyja rozwojowi nowotworów, chorób metabolicznych (np. cukrzyca typu 2), a także starzeniu. Rośnie zainteresowanie użyciem inhibitorów kinazy mTOR, a w szczególności analogów rapamycyny (rapalogów) jako leków.
Przy zbyt obfitej diecie organizm uruchamia mechanizmy, częściowo zależne od mTOR, które mają na celu gromadzenie składników odżywczych (na wypadek okresu ich braku). Może to prowadzić do otyłości czy chorób metabolicznych. TORC1 pośredniczy w aktywacji biosyntezy i akumulacji lipidów w adipocytach, a w konsekwencji również rozrostowi tkanki tłuszczowej. Chroniczna hiperaktywacja TORC1 w otyłości powoduje nie tylko anormalną lipogenezę w tkance tłuszczowej, wątrobie i mięśniach, ale również insulinooporność. Mechanizm ten opiera się o negatywne sprzężenie zwrotne zależne od S6K oraz IRS1 prowadzące do wyłączenia ścieżki zależnej od receptora insuliny, a w konsekwencji spadku wychwytu glukozy, glikogenezy, a wzrostu poziomu glukoneogenezy w komórkach wątroby. powyższe sprzężenie zwrotne S6K-IRS1 odgrywa znaczącą rolę w patogenezie hiperglikemii, hiperinsulinemii, a ostatecznie cukrzycy typu 2.
Mechanizmy zależne od aktywacji mTOR sprzyjają spontanicznemu rozwojowi nowotworów. eIF4E, aktywowany przez TORC1 stymuluje transkrypcję wielu pro-onkogenów, w tym regulatorów cyklu komórkowego czy białek anty-apoptotycznych (np. Mcl-1). Hamowanie autofagii w wyniku aktywacji mTOR również sprzyja rozwojowi chorób nowotworowych. TORC2 może również w sposób bezpośredni promować rozwój nowotworów poprzez aktywację kinaz z grupy AGC (AKT, SGK). Kinazy te promują proliferację, przeżycie komórki, a także wychwyt składników odżywczych.
Wiele dziedzicznych zespołów nowotworowych (np. stwardnienie guzowate), ma swoją genezę w mutacjach genów dla negatywnych regulatorów ścieżek zależnych od mTOR, jak np.: TSC1 (Tuberous Sclerosis 1), TSC2 (Tuberous Sclerosis 2), LKB1 (liver kinase B1, znany również jako STK11, Peutz-Jeghers syndrome), PTEN (phosphatase and tensin homolog deleted on chromosome ten, znany róenież jako MMAC1, mutated in multiple advanced cancers 1), NF1 (Neurofibromatosis) czy VHL (Von-Hippel Lindau). Onkogenne mutacje genów sieci sygnalnej PI3K-AKT, w sposób bezpośredni powiązanej z kompleksami TROC1 oraz TROC2 mogą również i często powodują rozwój nowotworów.
 źródło: [2] |
 źródło: [3] |
|---|
{kind=link}
{kind=link}
{kind=link}
Zobacz też
- mOTR w bazie NCBI
- mTOR w bazie RCSB PDB
- mTOR w bazie GeneCards
- Atlas of Genetics and Cytogenetics in Oncology and Haematology
Literatura
- ↑ Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. „Science”. 253. 5022, s. 905–9, 1991.
- ↑ 2,0 2,1 2,2 2,3 Laplante M, Sabatini DM. mTOR signaling at a glance. „Journal of Cell Science”. 122. 20, s. 3589-94, 2009.
- ↑ 3,0 3,1 3,2 Laplante M, Sabatini DM. mTOR signaling. „Cold Spring Harbor Perspectives in Biology”. 4. 2, s. a011593, 2012.
- ↑ Zheng XF, Florentino D, Chen J, Crabtree GR, Schreiber SL. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. „Cell”. 82, s. 121-130, 1995.
- ↑ Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. „Nature Reviews Molecular Cell Biology”. 10. 5, s. 307-318, 2009.
- ↑ Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. „Current Biology”. 19. 22, s. R1046-R1052, 2009.
- ↑ Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Heiden MGV, MacKeigan JP, Finan PM, Clish CB, Murphyemail LO, Manningemail BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. „Current Biology”. 39. 2, s. 171-183, 2010.
- ↑ Falcon BL, Barr S, Gokhale PC, Chou J, Fogarty J, Depeille P, Miglarese M, Epstein DM, McDonald DM. Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. „Cancer Research”. 71. 5, s. 1573-83, 2011.
- ↑ Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. „Journal of Biological Chemistry”. 284. 18, s. 12297-305, 2009.
- ↑ Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. „EMBO Journal”. 31. 5, s. 1095-108, 2012.
- ↑ Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. „Nature Reviews. Molecular Cell Biology”. 12. 1, s. 21-35, 2011.